Original
Articles
Role
of platelets in the pathogenesis of antiphospholipid syndrome
NK
Singh1*, Dibya R Behera2, DP Yadav3, A
Gupta3, D Dash4, D Bandyopadhyay5
Author Affiliations
1 Professor, Department
of Medicine & In-Charge Division of Rheumatology, Institute of Medical
Sciences (IMS), Banaras Hindu University (BHU), Varanasi, India
2 JR-3 in Medicine, IMS,
BHU, India
3
Ex-Residents in
Medicine, IMS, BHU, India
4
Professor,
Department of Biochemistry, IMS, BHU, India
5 Assistant Professor,
Department of Biochemistry, All India Institute of Medical Sciences,
Bhubaneswar, Orissa, India
*Correspondence:
Dr. N K Singh
IJRCI.
2013;1(1):OA2
Received:
12 January 2013, Accepted: 15 May 2013, Published: 16 July 2013
© IJRCI
Abstract
Aim
To delineate the role of platelets in thrombotic process in APS
patients.
Background
Pathogenesis of APS is an ongoing area of research and studying the
role of platelets will be helpful in developing newer diagnostic and
therapeutic strategies.
Materials and methods
Forty patients with APS, diagnosed as per modified 2006 Sapporo’s
Criteria and who were not on aspirin or any other antiplatelet drug, were
included. The same number of age- and sex-matched healthy controls was also
recruited for comparison. The following platelet function studies were
performed using the blood samples collected from APS patients as well as
healthy controls: platelet aggregation studies, platelet secretion of dense
granules (a. total degranulation b. platelet secretion of granules in relation
to time c. visualization of platelet degranulation), clot retraction studies,
and western blot studies on clot retracted samples for demonstration of
activated proteomes.
Results
A significant increase (P < 0.001) in the platelet aggregation in APS patients as compared to healthy controls was noted. The subjects also showed a significant increase (P < 0.05) in the platelet granule release as well as more degranulation (P < 0.001) in relation to time at stored condition, which were well-visualized under phase-contrast microscope. Sixty-five percent of APS patients showed lesser as well as delayed clot retraction as compared to healthy controls, signifying that the platelet clots are less retractile in APS patients.
Conclusion
The study clearly
demonstrates the hyperactivity of platelets in APS patients in each step of
their activation as compared to the controls. This indicates the major role
played by platelets in APS pathogenesis.
Introduction
Antiphospholipid antibody syndrome (APS) is a systemic autoimmune disorder characterized by increased tendency for arterial and/or venous thrombosis and adverse pregnancy outcome in the presence of various antiphospholipid antibodies.1 APS is classified as primary in the absence of another autoimmune disease such as systemic lupus erythematosus, and secondary if such disorders are present.2 Thrombosis is the most common clinical event in APS. The pathogenesis of APS has been described as multifactorial. Numerous pathological mechanisms have been proposed, including activation of endothelial cells, monocytes, and/or platelets; inhibition of natural anticoagulant pathways such as protein C, tissue factor inhibitor, and annexin V; activation of complement system; and impairment of fibrinolytic system.3, 4 However, the exact pathogenesis is still inconclusive. One model had drawn a hypothesis parallel with the thrombosis of heparin-induced thrombocytopenia (HIT) suggesting the possible existence of a pathogenic interaction between antiphospholipid antibodies (aPL) and platelets.5
Earlier studies have proposed hyperactivity of platelets as an important mechanism responsible for hypercoagulable state in APS. But very limited studies have assessed the platelet function in APS. Forastiero et al. showed that the presence of increased urinary levels of 2, 3 dinorthromboxane B2, a stable thromboxane metabolite, is an important indicator of activated circulating platelets in APS patients.6 The study finding confirms a similar observation of plasma and urine levels of β-thromboglobulin, a protein that is released from platelet storage organelles upon activation.7 Fanelli et al. demonstrated a significant increase in the expression of CD62p, an activation marker on platelet, in APS patients.8 Most of these studies have used surrogate markers for evaluating platelet functions (mostly degranulation and/or surface expression of protein on activation). These surrogate markers may not completely reflect the functional status of platelets.
We hypothesize that there will be functional alteration in the platelets in patients with APS. In the present study, we have examined the in vitro platelet activation in patients with APS using several methods, since it has been recognized that platelet activation is a complex process and measuring single marker for activation alone may limit the ability to detect platelet activation.
Patients and methods
The study was carried out on the patients attending Departments of Medicine, Rheumatology, Nephrology, and Obstetrics and Gynaecology in our institute. Patients diagnosed to have APS, as per the modified 2006 Sapporo’s Criteria, and who were not on aspirin or any other antiplatelet drug/heparin/oral anticoagulants were enrolled in the study.9 Age- and sex- matched healthy controls were also recruited. The study was approved by the Institute Ethical Committee and informed consent was taken from the patients.
Serological
tests
Standard immunological tests were carried out to estimate different autoantibodies namely ANA, Anti-dsDNA, rheumatoid factor (RF), IgG and IgM anticardiolipin antibodies (IgG aCL and IgM aCL), and lupus anticoagulant (LA). The presence of aCL of IgG and IgM isotypes was measured by an enzyme-linked immunosorbent assay (ELISA) using ORGENTEC diagnostic kit (ORGENTEC Diagnostika GmbH, Germany). LA activity was detected by coagulation assays, adhering to the guidelines of the International Society on Thrombosis and Hemostasis (ISTH) using TULIP kit (Tulip Diagnostics, India). Repeat testing of LA, IgM and IgG ACLA were done at least 12 weeks apart. However, antibody to β2 glycoprotien1 was not carried out due to non-availability of this test kit during the study period.
Platelet
function studies
Blood samples of 10 ml were collected using standardized atraumatic protocol from the antecubital fossa following proper antisepsis precautions. First few drops of blood were discarded and the rest were collected in plastic tubes containing freshly prepared 1.4 ml of citrate-phosphate-dextrose anticoagulant (CPDA). Platelet rich plasma (PRP) was separated from the samples and were analysed within 3 hours. The laboratory personnel were blinded to the source of samples.
The following platelet function studies were performed:
1. Platelet aggregation studies
2. Studies pertaining to platelet secretion of dense granules, which comprised of :
· Total degranulation
· Platelet secretion of granules in relation to time
· Visualisation of platelet degranulation and alteration of its morphology by phase contrast microscopy
3. Clot retraction studies by tube method
4. Western blot studies on clot retracted samples for demonstration of activated proteomes
All the above tests were performed simultaneously in blood samples collected from APS patients as well as healthy controls to avoid bias. Details of these procedures and their interpretation are as follows:
1. Platelet aggregation studies
Platelets were activated (non-stirring) and aggregated under stirring condition (12,000 rpm) at 37°C in a Chronolog platelet ionized calcium aggregometer (model 600) by thrombin (0.5 U/ml). Aggregation was measured as the percentage change in light transmission, where 100% refers to the transmittance through blank sample.10 The principle employed here is that when platelets suspended in buffer aggregate, the solution becomes clearer and transmittance of light increases, which is evaluated using the calcium aggregometer.
2. Study pertaining to platelet secretion
(degranulation) of dense granules
Platelets were stimulated with thrombin (0.5U/ml) for 3 minutes. Activated (unstirred) and aggregated (stirred) platelet samples were centrifuged at 800g for 1 min along with 1 mM EDTA to prevent further platelet activation. The supernatants obtained were added to reaction mixture containing luciferin-luciferase in microplates (Lumitrac 200, Greiner). Luminescence generated due to the reaction of released adenine nucleotides was read in a luminescence microplate reader (BioTek, model FLx800TBI).10
3. Clot retraction studies by tube method
During western blot analysis of clot-retraction samples, fibrin clot retraction was studied by adding procured fibrinogen at 2mg/ml to the washed platelets to eliminate the interference of plasma proteins.11, 12
In order to perform fibrin clot retraction assay, the experimental method described by Osdoit and Rosa was essentially followed.12 Washed platelets (0.6 × 109 cells/ml) were incubated with Ca2+ for 10 minutes. Fibrinogen (2 mg/ml) was added to the resuspended platelets thereafter. Fibrin clot retraction was initiated by the addition of thrombin (1 IU/ml). The clot was allowed to retract for 60 min at 37°C. In control experiments, retraction was prevented by the omission of calcium in the reaction mixture. Clots were lysed by boiling with Laemmli buffer for 20 min and stored at 20°C, until further analysis was carried out. Clot retraction was assessed by analyzing the digital photos with ImageJ 1.36b software (NIH, USA). The extent of retraction was expressed as a percentage of retraction defined as [1- (area t/area t0) x 100], where area t0 was the area occupied by the fibrin clot in the absence of platelets (‘negative control’) and ‘t’ denoted the area occupied by the retracted fibrin clot.13
4. Western blot studies on clot retracted
samples for visualizing activated proteomes
For analysis by western blotting, clots were lysed by the addition of one-fourth volume of 5× sample lysis buffer (1× contained 0.06 M Tris HCl pH 6.8, 2% SDS, 5% v/v 2-mercaptoethanol, 10% glycerol, 0.016% bromophenol blue, and 1mM sodium orthovanadate) and were heated at 90°C in a dry bath for 20 min. Platelet proteins were separated on 10% or 10-18% gradient SDS-PAGE as needed, and were electrophoretically transferred to polyvinylidene difluoride membranes (Pierce Biotechnology, Rockford, IL, USA or Bio-Rad Laboratories, CA, USA) using Nova Blot semidry system (Multiphor II & EPS 600, Amersham Pharmacia Biotech), as per the manufacturer’s instructions. Blots were then incubated for 2h or overnight at 4°C with different dilutions of primary antibody (mouse anti-phosphotyrosine with1:1000 dilutions in 2% bovine serum albumin in 1×tris-buffered saline with tween). The blots were incubated for 2h with horseradish peroxidase-labeled secondary antibody (anti-mouse IgG) after washing. The antibody binding was detected using enhanced chemiluminescence and bands were densitometrically quantified by Agfa Duoscan T1200 flatbed scanner and GeneTools software (Syngene, UK).10
Statistical
analysis
Standard statistical methods were used. Parametric methods (t test) were employed for evaluation and P < 0.05 (2- tailed tests) was considered as significant. Data were presented as means±SD of all individual experiments from different blood samples. Immunoblots shown were representatives of at least three different experiments.
Results
A total of 40 APS patients (20 each of primary and secondary APS) were included in the study. All the 20 secondary APS patients had SLE. The table 1 depicts the demographic data of all the enrolled subjects.
Table 1: Demographic data of APS
patients and control groups
|
Study groups |
Number of subjects |
Age (years) Mean±SD |
Female/male |
Thrombosis* |
Smoking history present/ absent |
Medication history# |
|
APS
Patients |
||||||
|
Total |
40 |
26.75±6.49 |
33/7 |
Yes |
4/36 |
No |
|
Primary |
20 |
27.3±5.53 |
16/4 |
Yes |
2/18 |
No |
|
Secondary |
20 |
26.15±7.4 |
17/3 |
Yes |
2/18 |
No |
|
Control group |
||||||
|
Healthy controls |
40 |
27.82±5.68 |
35/5 |
No |
5/35 |
No |
* Venous/arterial thrombosis
# Antiplatelet drugs including aspirin,
and oral anticoagulants or heparin
Evaluation of the immunological parameters revealed moderate to high titers (> 40 GPLU/ ml) of IgG and IgM anticardiolipin antibody in 31 (77.5%) and 14 (35%) patients respectively, whereas the presence of both the antibodies were reported in 10 (25%) patients. Lupus anticoagulant was present in 28 (70%) patients. Eighteen (45%) patients had both anticardiolipin and lupus anticoagulant antibodies.
Platelet
aggregation studies
The results of platelet aggregation studies, conducted on both APS patients and healthy controls, are given in table 2. A significant increase (P < 0.001) in the platelet aggregation was seen in APS patients when compared to healthy controls. Aggregation pattern was found to be similar in all the cases (n=40) studied.
Table 2: Platelet aggregation in
healthy controls and APS patients in relation to time
|
Study groups |
Number of subjects |
Aggregation (light
transmittance) (Mean±SD) |
||
|
1 min |
2 min |
3 min |
||
|
Healthy controls |
40 |
30.12±2.18 |
60.65±9.85 |
65.02±13.52 |
|
APS patients |
40 |
50.23±4.58 |
75.01±10.56 |
77.77±11.72 |
|
P-value |
< 0.001 |
< 0.001 |
< 0.001 |
|
Studies
pertaining to platelet secretion of dense granules
(a) Total degranulation of dense granule
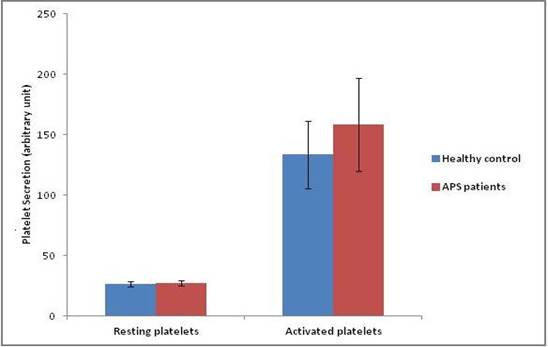
APS patients showed significant increase (P < 0.05) in the release of platelet granules when compared to the controls upon using the same agonist for activation at the same concentration (thrombin= 0.5U/ml) (n=20). The release of the dense granules from the platelets was found to be more prompt and greater in APS patients than the ‘control’ platelets (Fig 2).
Fig 2: ATP/ADP release of
resting and activated platelets in healthy controls and APS patients
(b) Platelet secretion of granules with time
The platelet degranulation was monitored at 0, 1, 3 and 5-hour time points under storage condition. Increase in the number of platelets was noted in both APS patients as well as in controls, but it was significantly more (P < 0.001) in APS patients (Table 3). These findings were further substantiated by the illustration of platelet degranulation process using phase contrast microscopy.
Table 3: Platelet degranulation
in relation to time in APS patients as compared to healthy controls
|
Study groups |
Number of subjects |
Platelet degranulation (Mean±SD) |
|||
|
At rest |
1 hour |
3 hours |
5 hours |
||
|
Healthy controls |
20 |
25.55±2.48 |
35.25±4.79 |
52.30±8.41 |
66.10±7.20 |
|
APS patients |
20 |
25.90±2.42 |
44.85±5.50 |
60.80±7.01 |
80.05±9.01 |
|
P-value |
0.654 |
<0.001 |
<0.001 |
<0.001 |
|
(c)
Microscopic
visualization of platelet degranulation
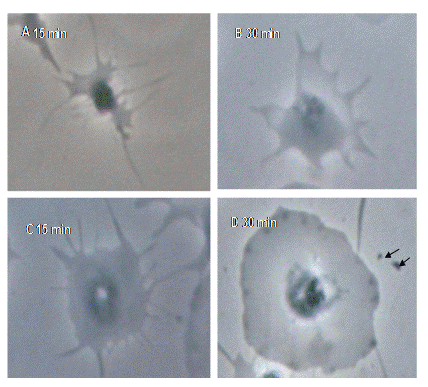
The platelet degranulation was evaluated using phase-contrast
microscopy (Fig 3). The arrows in figure 3 indicate an exaggerated
degranulation response seen after the addition of thrombin agonist in APS patients.
The occurrence of degranulation was concluded from morphologic appearance of
spread platelets in multiple samples. Similar morphologic pattern was present
in all the 20 specimens obtained from APS patients. Thus, the change in
platelet shape associated with the degranulation process was evident on
microscopic evaluation.
Fig 3: Alteration in platelet
morphology and degranulation in response to thrombin addition in healthy
controls (A & B) and APS patients (C &D), read at 15 and 30 minutes.
The C and D slides show the exaggerated degranulation response noted after the
addition of thrombin agonist (marked by the arrows in slide D).
Clot
retraction studies
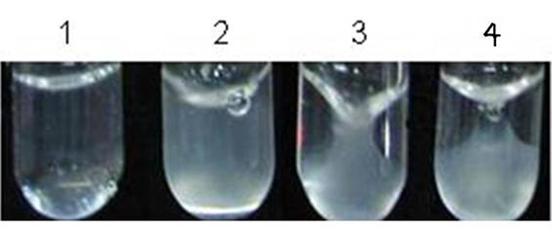
Clot retraction studies were carried out using APS samples (n=20) in comparison to the control group and also using platelet-poor plasma. Mild clot retraction was observed in 13 (65%) APS patients when compared to the healthy controls (Fig 4). The clot retraction was moderate in 5 (25%) APS patients and in 2 (10%) patients, it was comparable to that of the controls. Hence it was concluded that majority of APS patients (65%) showed less clot retraction in contrast to healthy controls.
Fig 4: Tube 1 containing platelets, Ca2+, and fibrinogen did not show
clot formation, as it was devoid of the agonist (thrombin). Tube 2 contained
fibrinogen and thrombin, but was devoid of Ca2+ and platelets. Addition of
thrombin catalyzed the polymerization of the fibrinogen to fibrin. Although the
clot formation had occurred, it did not retract on standing, suggesting that
the clot retraction phenomenon is solely due to platelets supported by Ca2+.
Tube 3 had ‘control’ platelets, Ca2+, fibrinogen, and thrombin. It showed
severe clot retraction at 30 minutes. Tube 4 had ‘APS’ platelets, Ca2+,
fibrinogen, and thrombin. Less clot retraction than the control was noted (Tube
3) at 30 min.
Western blot of the clot
retraction samples
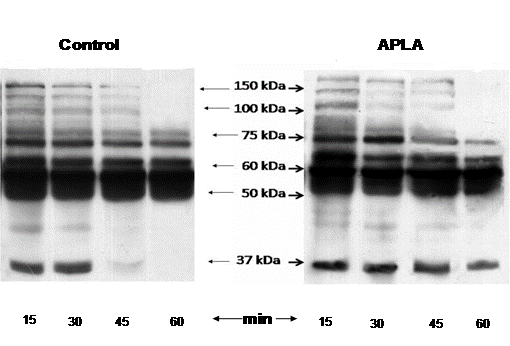
Dephosphorylation of proteins in the clot retraction samples was found to be higher in control than the APS platelets. This could probably explain why the clot retraction was more in the former group. Western blotting with anti-phosphotyrosine antibody (anti-PY) demonstrated an additional band at around 37 kDa in APS clot retracted samples (n=10) at the time points 15, 30, 45, and 60 minutes (Fig 5).
Fig 5: Anti-PY western blot of
clot retracted samples of APS and control. It demonstrated activity of new
proteins formed in APS patients (containing phosphotyrosine) in response to thrombin
at 15, 30, 45, and 60 minutes. The blot also showed inactivity of the platelets
with time.
Discussion
Platelet activation may play an important role in the thrombosis associated with APS. Numerous studies have investigated platelet activation in APS patients, and the measurement of platelet release products such as β- thromboglobulin was the main parameter considered by most of the earlier studies for the assessment.7 Platelet degranulation results in the expression of CD62p (P-selectin) after α-granule release and CD63 on the platelet surface membrane, which is followed by lysosomal and dense granule secretion.14, 15, 16 Study conducted by Joseph et al. in 20 primary APS patients reported a significant increase in median platelet CD63 expression and plasma soluble P-selectin.17 Due to the complexity of the platelet activation process, measuring the surrogate degranulation markers alone may limit the ability to detect platelet activation. Therefore, in the current study, all steps of platelet activation were studied to elucidate the role of platelets in the pathogenesis of APS.
Urbanus et al. showed that the interaction of aPL with platelets can occur in at least three different ways: 1. Immunoglobulins may bind through the Fab fragment with specific platelet antigens in a classic antigen-antibody reaction, 2. Immune complexes may bind to platelets via FcgRII receptor, and 3. aPL, like other immunoglobulins, may bind to platelets in a non-specific manner by mechanisms speculated to involve platelet membrane injury.18 Shi et al. observed that human anticardiolipin (aCL) binds to platelets in a β2GPI-dependent way.19 The only FcgR molecules present on platelets are the FcgRII. Activation of the FcgRII receptor causes platelet activation and granule release.
In concurrence to the previous study findings, the present study also demonstrated increased aggregability of platelets derived from APS patients when compared to the control group (P=0.001). Wiener et al. showed significant spontaneous platelet aggregation in 21 patients of SLE with APS.20 The two mechanisms indicating that the platelet aggregation and secretion are highly connected events are: 1) Activated platelets degranulate that can either induce or mediate platelet aggregation and 2) Platelet-platelet contact during aggregation can lead to activation of certain pathways that promote secretion of platelet granules.21, 22 Furthermore, platelet dense granule deficiency (Hermansky-Pudlak syndrome) is associated with defective platelet aggregation in vitro and bleeding diathesis.23 As hyperaggregability alone is not a marker of hyperactivity, we studied platelet release reaction, mainly dense granule (rich in ADP/ATP).
In the present study, we measured adenine nucleotides as a marker of degranulation and the study findings showed that platelets are hyperactive even during release reaction (P-value= 0.024). The result was comparable to the study by Joseph et al., who used CD62p (P-selectin) expression as a marker of degranulation (P-value=0.007 for APS vs. control).17 It clearly demonstrated that, at room temperature (37˚C), platelets separated from APS patients demonstrated more secretory activity when compared to healthy controls. But, the study did not show the effect of time on platelet secretion indicating that it is not apparent whether platelets of APS patients maintain the hypersecretory state for long as compared to control. Hence, we evaluated platelet release reaction in relation to time and it was found that their secretion gradually increased with time, which was significantly more than that of the healthy subjects’ platelets. Visualization of degranulation of platelets under the microscope clearly showed the hyperactivity of platelets in APS patients. Additionally, exaggerated response of platelets caused rapid changes in shape and degranulation. The above observation substantiates the previous studies suggesting that the platelets from APS patients are hyperactive.
As part of platelet function studies, we also carried out clot retraction study and found that clots formed of platelets from APS patients are less retractile than controls. Thirteen (65%) out of 20 patients showed much less clot retraction as compared to healthy controls. However, there is no evidence available from previous studies to compare these results. As per the proposed mechanism, platelets during activation form many proteins that are active due to the presence of phosphotyrosine moiety. Western blot clearly showed that platelets from APS patients have more of the tyrosine phosphorylated proteins than controls. With time, the dephosphorylation of proteins in the clot retracted samples in ‘control’ platelets was found to be higher than the APS platelets. This could be the probable explanation for the occurrence of increased clot retraction in the ‘control’ platelets than the APS platelets. The lesser retractility of platelets in APS could help aggravating the thrombotic occlusion.
Limitation of the present study is that it did not project the in vivo picture of thrombosis in APS. Future research is aimed at investigating the role of platelet-derived microparticles (in distinction from macrophage-derived microparticles) in APS patients. As microparticles are known to be highly thrombogenic, the evaluation may help to further explore the thrombotic mechanism in APS.
In summary, we demonstrated that the platelets are hyperresponsive in APS patients in comparison to age matched controls. They are hyperaggreagable, hypersecretory (hyper-degranulating), showing increased degranulation with massive shape change, and having less clot retracting power as compared to control platelets. All these features clearly signify that the hyperfunction of platelets play a major role in the pathogenesis of APS.
Competing interests
The
authors declare that they have no competing interests.
References
1.
Levine
JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002;
346(10):752–63.
2.
Alarcon-Segovia
D, Deleze M, Oria CV, et al..
Antiphospholipid antibodies and the antiphospholipid syndrome in systemic lupus
erythematosus: a prospective analysis of 500 consecutive patients. Medicine
(Baltimore). 1989; 68(6): 353-65.
3.
Arnold
J, Holmes Z, Pickering W, Farmer C, Regan L, Cohen H. Anti-beta 2 glycoprotein
1 and anti-annexin V antibodies in women with recurrent miscarriage. Br J
Haematol 2001; 113(4):911–14.
4.
Rand
JH, Wu XX, Andree HA, Lockwood CJ, Guller S, Scher J, et al.. Pregnancy loss in the antiphospholipid-antibody syndrome—a
possible thrombogenic mechanism. N Engl J Med 1997; 337(3):154–60.
5.
Arnout
J. The pathogenesis of the antiphospholipid syndrome: a hypothesis based on
parallelisms with heparin-induced thrombocytopenia. Thromb Haemost. 1996;
75(4): 536-41.
6.
Forastiero
R, Martinuzzo M, Carreras LO, Maclouf J. Anti-beta2 glycoprotein I antibodies
and platelet activation in patients with antiphospholipid antibodies:
association with increased excretion of platelet-derived thromboxane urinary
metabolites. Thromb Haemost 1998; 79 (1): 42–45.
7.
Galli
M, Cortelazzo S, Viero P, Finazzi G, de Gaetano G, Barbui T. Interaction
between platelets and lupus anticoagulant. Eur J Haematol 1988; 41(1): 88–94.
8.
Fanelli
A, Bergamini C, Rapi S, Caldini A, Spinelli A, Buggiani A & Emmi L. Flow
cytometric detection of circulating activated platelets in primary
antiphospholipid syndrome. Correlation with thrombocytopenia and
anticardiolipin antibodies. Lupus 1997; 6(3): 61-67.
9.
Miyakis
S, Lockshin MD, Atsumi T, et al..
International consensus statement on an update of the classification criteria
for definite antiphospholipid syndrome (APS). J Thromb Haemost
2006;4(2):295–306.
10.
Shrivastava
S, Bera T, Singh SK, Singh G, Ramachandrarao P, Dash D. Characterization of
Antiplatelet Properties of Silver Nanoparticles. ACS Nano. 2009;3(6): 1357-64.
11.
Law
DA, DeGuzman FR, Heiser P, et al..
Integrin cytoplasmic tyrosine motif is required for outside-in IIb3 signalling
and platelet function. Nature. 1999;401(6755):808-11.
12.
Osdoit
S, Rosa JP. Fibrin clot retraction by human platelets correlates with IIb3
integrin-dependent protein tyrosine dephosphorylation. J Biol Chem. 2001; 276:
6703-10.
13.
Podolnikova
NP, Yakubenko VP, Volkov G L, Plow EF, Ugarova TP. Identification of a Novel
Binding Site for Platelet Integrin _IIb_3 (GPIIbIIIa) and α5β1 in the
ϒCDomain of Fibrinogen. J. Biol. Chem. 2003; 278: 32251-58.
14.
Stenberg
PE, Mc Ever RP, Shuman MA, Jacques YV, Bainton DF. A platelet al.pha-granule membrane protein (GMP- 140) is expressed on the
plasma membrane after activation. Journal of Cell Biolog. 1985; 101(3): 880-86.
15.
Berman
CL, Yeo EL, Wencel-Drake J, Furie BC,
Ginsberg MH, Furie B. A platelet al.pha-granule
protein that is associated with the plasma membrane after activation:
characterization and subcellular localisation of platelet-activation dependent
granule-external membrane protein. Journal of Clinical Investigation. 1986;78:
130-37.
16.
Nishibori
M, Cham B, McNicol A, Shalev A, Jain N, Gerrard JM. The protein CD63 is in platelet
dense granules, is deficient in a patient with Hermansky±Pudlak syndrome, and
appears identical to granulophysin. Journal of Clinical Investigation.1993;
91(4): 1775-82.
17.
Joseph
JE, Donohoe S, Harrison P, Mackie I.J, Machin SJ. Platelet activation and turnover
in the primary antiphospholipid syndrome. Lupus.1998; 7(5): 333-40.
18.
Urbanus
RT, Derksen RH, de Groot PG. Platelets and the antiphospholipid syndrome. Lupus
2008; 17(10):888.
19.
Shi
T, Giannakopoulos B, Yan X, et al..
Anti-beta2-glycoprotein I antibodies in complex with beta2-glycoprotein I can
activate platelets in a dysregulated manner via glycoprotein Ib-IX-V. Arthritis
Rheum 2006; 54(8): 2558-67.
20.
Wiener
HM, Vardinon N, Yust I. Platelet antibody binding and spontaneous aggregation
in 21 lupus anticoagulant patients. Vox Sanguinis.1991; 61(2):111-21.
21.
Siess
W. Molecular mechanisms of platelet activation. Physiol Rev. 1989;
69(1):58-178.
22.
Fox
JE. The platelet cytoskeleton. Thromb Haemost. 1993; 70(6):884-93.
23.
Zucker-Franklin
D. In Greaves MF, Grossi CE, Marmot AM, Zucker-Franklin D (eds). Atlas of Blood
Cells, Function, and Pathology. Vol. 2. Philadelphia: Lea & Febiger, 1988.